



FDA 's Process of Drug and Vaccine Evaluation, Part I
This will be a 3 part series. Part 2 will focus on how adverse event causality assessments should be made. Part 3 will reveal how the WHO corrupts the process. for vaccines.

Since President-elect Trump chose Robert F Kennedy, Jr. for the role of Health and Human Services (DHHS) secretary, there has been a lot of talk about vaccine safety. Are vaccines safe? How have they been evaluated? Would Mr. Kennedy revoke the licenses of vaccines?
Mr. Kennedy has said he is all for free choice, and that people who want a vaccine can get one. But at the same time, the DHHS is responsible for evaluating whether products it licenses are a) safe, b) effective and c) were manufactured properly. That is all FDA has to do with respect to drugs (and vaccines and biologics are considered a subclass of drugs): assure that each licensed product is safe, effective and produced under Good Manufacturing Processes standards.
Yet there are many conflicts of interest that may influence licensing decisions. Here are some examples:
· Members of Congress have tried to influence FDA decisions on drugs and medical devices.
· FDA employees often leave to work for companies whose products they regulated.
· In fact, FDA's lax rules seem to encourage this practice.
· Manufacturers pay FDA to review their products, and pay extra for a speedy review. Thus, FDA has come to see the pharmaceutical industry as its primary client, rather than the consumer.
Even FDA's appointed experts are sometimes disgusted by licensing decisions made by FDA brass, decisions that ignore the advice of the experts and of FDA staff--and they publicly quit.
Evaluations of the manufacturing process are inadequate due to insufficient inspection staff. FDA now accepts the results of inspectors working for others nations. FDA performed less than one third of the domestic inspections in 2022 than it performed in 2005. The Government Accountability Office advised FDA last month to find a way to retain inspection staff. However, FDA instead plans to do virtual inspections in future, via videocam, which are highly unlikely to turn up problems that manufacturers are trying to hide.
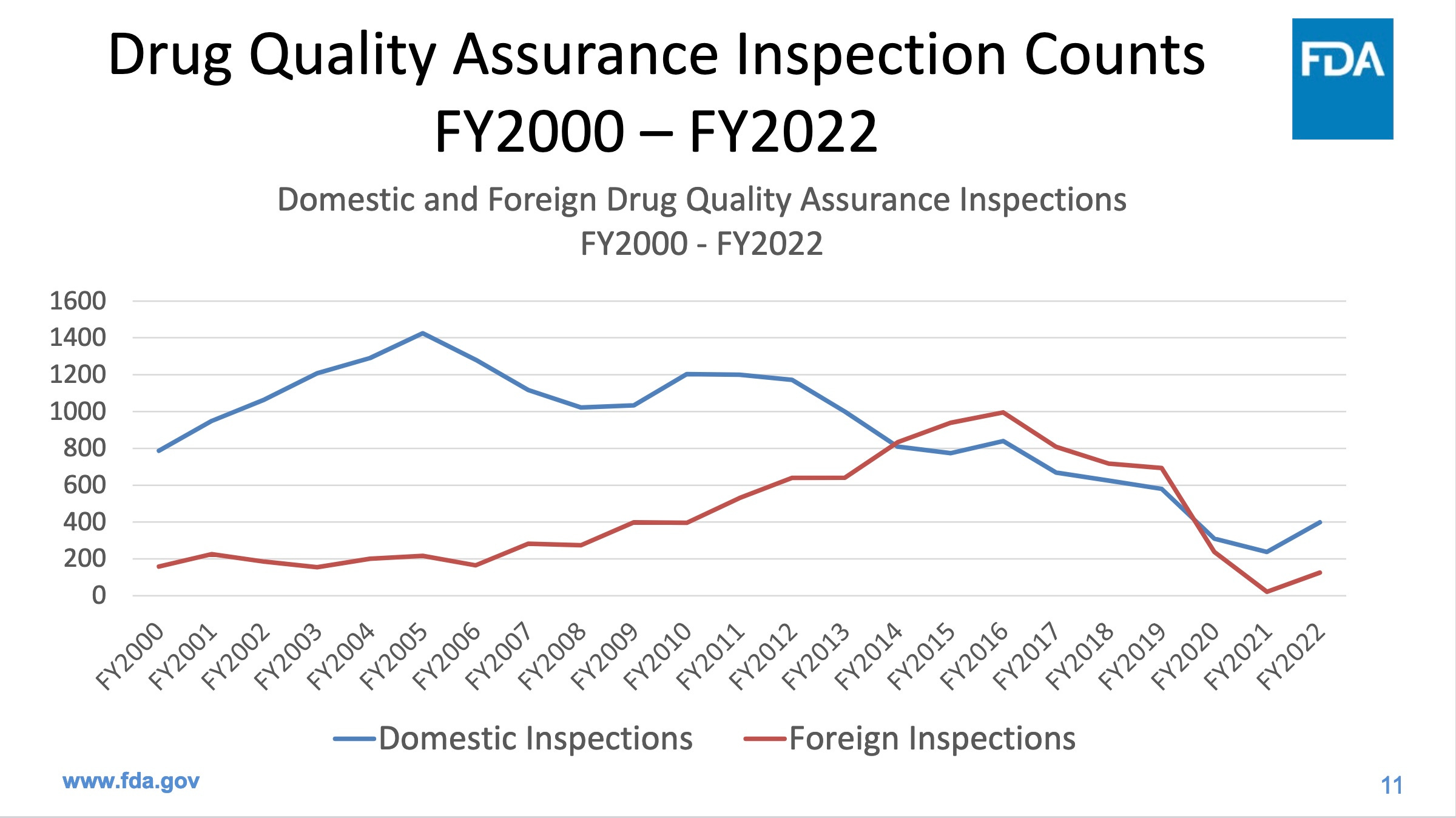
https://www.fda.gov/media/172786/download
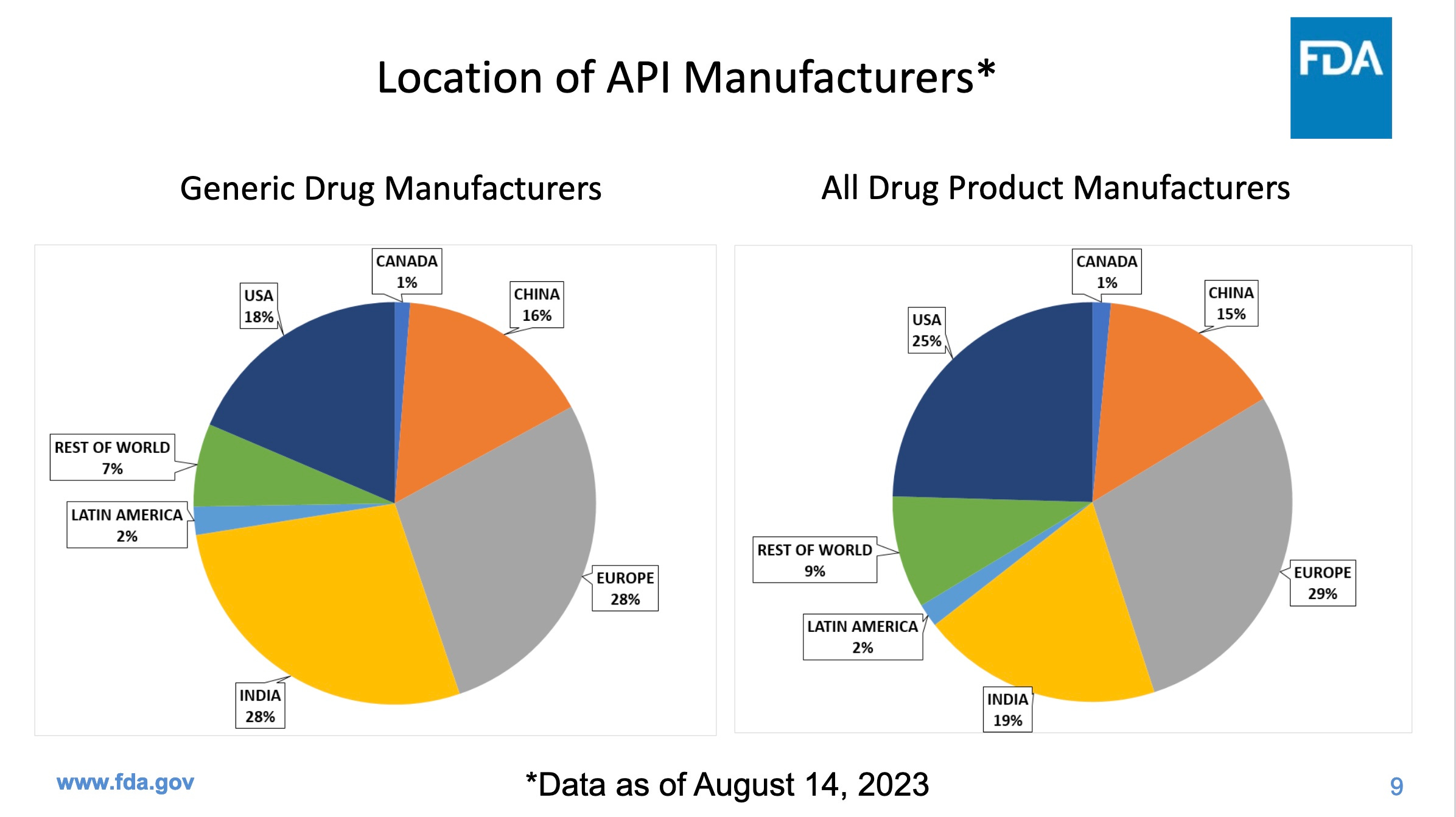
Inspections are particularly important in places like India and China, which together produce a whopping 44% of our generic drugs, but fail our inspections more often than US companies. The next two graphics come from the same FDA presentation as the first graphic.
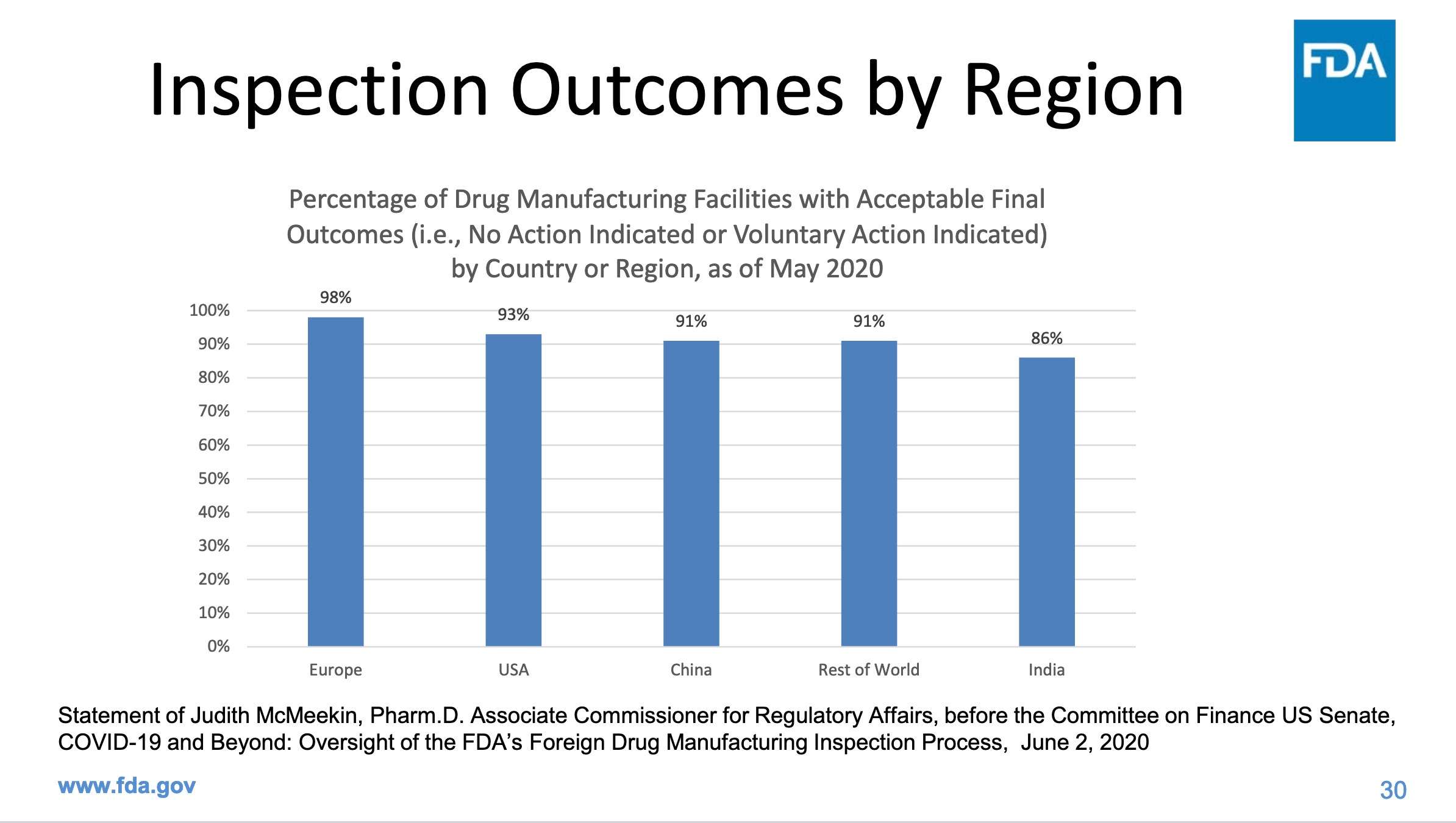
Note that the percentages of companies that passed FDA inspections refers to final outcomes: after the company has had a chance to fix the problems identified in an FDA inspection. FDA does not tell us how many failed the initial inspection. It is worrying that 1 in 7 manufacturers in India still can’t get things right even after their flaws were identified, and that the same is true for China, with 1 in 11 failing final inspections. In the US, 1 in 14 fail final inspections. How many of us have been consuming drugs produced by these companies?
Given the history of problems with FDA's evaluations and licensing decisions, it is only prudent that Mr. Kennedy make public the data FDA has collected and analyzed regarding the safety, efficacy and quality of all its licensed drugs, biologics (which includes vaccines) and medical devices.
In 1972, when the regulation of vaccines was shifted to FDA from NIH, FDA was required to review every vaccine license and guess what? About half the licenses were voluntarily surrendered or revoked! Maybe it is time to do something similar with the many licensed but questionable products.
FDA actually buys data from health insurance companies, HMOs and drugstore companies. It has data on over 100 million Americans to evaluate the safety of drugs and vaccines. In the vaccine and biologics arena alone, FDA is allotted a staff of 18 officials (and unknown numbers of underlings) to study the safety of these products. We have seen very little of their data and their conclusions. Almost since the start of COVID, this small agency has been missing 7 of its 18 officials. It is missing 39% of its supervisory staff!
It also changed its name from the Office of Biostatistics and Epidemiology to the Office of Biostatistics and Pharmacovigilance on October 30, 2024, in an apparent effort to become harder to find, or perhaps to appear vigilant.
Yet in the last four years, under Biden, DHHS has increased its total staffing by 19%. Is the staffing shortage in the one FDA office that is tasked with studying post-licensure vaccine safety data a deliberate attempt to minimize studying the safety of the COVID vaccines?
By law, all the data that FDA reviewed for licensed products pre-licensure should come into the public domain following licensure. But it often does not. Citizens and non-profit organizations must routinely file lawsuits against FDA to obtain the data we are entitled to see.
A recent example is the case of Del Bigtree's Informed Consent Action Network (ICAN) non-profit, which has had to take FDA to court to get release of the Pfizer licensing documents, and then had to go to court again because FDA illegally withheld a million pages. This process is very expensive, and it has taken 3 years (so far) to obtain about 60% of the relevant materials.
Another example is ICAN's attempt to obtain the safety data used to license the IPOL polio vaccine. Despite claims the vaccine was properly evaluated by FDA, the data provided by FDA show only three days of safety assessment following vaccinations. This is entirely inadequate to assess for serious adverse events, which may develop weeks or longer after vaccination. So in 2022 ICAN asked FDA to withdraw the vaccine for infants and children until a clinical trial with adequate safety data was conducted.
This week the NY Times got hold of this story and tried to make it about Robert F. Kennedy, Jr. revoking the polio vaccine, even though he is not a part of the lawsuit and has never asked for the vaccine to be revoked. The Times used the fact that Aaron Siri, ICAN's lead attorney, also sometimes works with Kennedy.
So, let's look at how vaccines and drugs are assessed for safety. Every drug or vaccine may cause a variety of different adverse effects. Usually, these adverse effects cannot be predicted in advance, despite our knowledge of pharmacology and human biology. Nor can we predict who might be most likely to suffer an adverse event, in most cases.

Therefore, after a drug has been designed, a staged process is conducted, first with extensive animal experiments, to see how the drug is metabolized and its effects on experimental animals. If an FDA review suggests the studies are adequate, an Institutional Review Board (IRB) must approve the initiation of human studies, and then they can proceed. The concept of IRBs dates back to 1974, following revelations about the government's own unethical Tuskegee syphilis study--when the federal government finally required an independent safety review before research using human subjects could be initiated.
IRBs initially were all associated with universities and research centers that received federal funding. But subsequently commercial IRBs were allowed, which will review your human subject experiment for a fee. With the advent of commercial IRBs, we can assume that a drug manufacturer will be able to find an IRB somewhere that will approve almost anything.
Once there is IRB approval, human experiments can start. There are 3 phases before a drug can be presented for licensure, and FDA needs to approve the move to Phase 2 and Phase 3 after reviewing the data from the previous phase.
According to FDA, Phase 1 studies usually involve 20-80 people and their intent is to see whether the drug at different doses is harmful. Phase 1 subjects do not benefit in any way from being subjects, because no one has yet determined whether the drug has efficacy for anything. Phase 2 studies involve from several dozen to 300 subjects and evaluate both safety and efficacy. Phase 3 studies employ between several hundred and several thousand subjects.
There may also be Phase 4 studies, or postmarketing studies. In this case, FDA needs more information about the product but is willing to license it. FDA requires that the manufacturer continue to collect data on the drug, under specific circumstances, to satisfy additional demands. These studies may go on for a number of years, during which the efficacy or safety of the product (at least for certain demographics) remains in doubt.
Here is an example of one (of many) post-marketing requirements FDA applied to Pfizer/BioNTech's COVID vaccine on August 23, 2021:
https://www.fda.gov/media/151710/download
"POSTMARKETING REQUIREMENTS UNDER SECTION 505(o) Section 505(o) of the Federal Food, Drug, and Cosmetic Act (FDCA) authorizes FDA to require holders of approved drug and biological product applications to conduct postmarketing studies and clinical trials for certain purposes, if FDA makes certain findings required by the statute (section 505(o)(3)(A), 21 U.S.C. 355(o)(3)(A)).
We have determined that an analysis of spontaneous postmarketing adverse events reported under section 505(k)(1) of the FDCA will not be sufficient to assess known serious risks of myocarditis and pericarditis and identify an unexpected serious risk of subclinical myocarditis.
Furthermore, the pharmacovigilance system that FDA is required to maintain under section 505(k)(3) of the FDCA is not sufficient to assess these serious risks. Therefore, based on appropriate scientific data, we have determined that you are required to conduct the following studies:
4. Study C4591009, entitled “A Non-Interventional Post-Approval Safety Study of the Pfizer-BioNTech COVID-19 mRNA Vaccine in the United States,” to evaluate the occurrence of myocarditis and pericarditis following administration of COMIRNATY. We acknowledge the timetable you submitted on August 21, 2021, which states that you will conduct this study according to the following schedule: Final Protocol Submission: August 31, 2021 Monitoring Report Submission: October 31, 2022 Interim Report Submission: October 31, 2023 Study Completion: June 30, 2025 Final Report Submission: October 31, 2025"
FDA gave BioNTech 4 years and 2 months to complete this study, knowing that the vaccine's makeup would be entirely different than it was in 2021, because FDA was about to recommend booster doses using a different formulation. In fact, there have been 4 US COVID/BioNTech formulations, and so the study population will consist of subjects receiving a variety of different vaccines. Clearly, this is not an ideal way to study a vaccine.
Also, FDA knew that everyone who wanted to be vaccinated would almost certainly have been vaccinated by 2025, so the results would have very little effect on vaccination decisions, when they finally became available.
However, by ordering that this study and others be conducted, FDA was protecting itself from charges that it was uninterested in myocarditis as a vaccine side effect--and also FDA claimed it was simply unable to make an assessment regarding myocarditis using VAERS reports by themselves. (This is untrue: the myocarditis reports from young males are up to 100 times higher than expected, allowing FDA to make accurate determinations of the mRNA COVID vaccines' danger to young males, and to a lesser extent young females.)
Now that I have explained the overall process of how FDA evaluates and approves drugs, I will go into depth on how safety is assessed in Part II. In Part III, we will see how the WHO corrupts that process.









Are vaccines safe? The answer to the question: NO! Those who promote vaccines have to be ignorant programmed robots. The history of vaccines from the beginning showed they left a wake of injury and death behind them. If medical doctors had studied nutrition, they would be aware that the only thing that belonged in the bloodstream would be nutrients from food. Natural immunity is strengthened by nutrients...not foreign proteins, elements or chemicals. Anything other than nutrients which feed the organs and cells would injure or destroy the human body.
As always, a huge thanks to Dr. Nass. And I greatly hope Congress confirms RFK JR.'s nomination but I'm not optimistic.